

What Happened In The MassTorts World Last Week? 2018-Jun-11
Stryker Alert: LFIT V40 Hip Replacement Parts Unsafe for Use
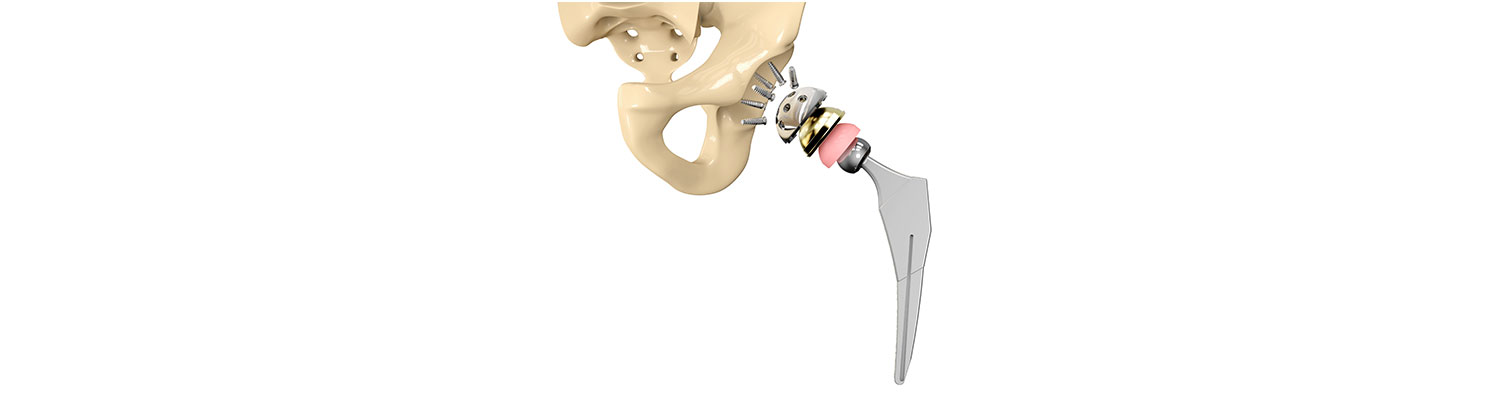
A safety alert by hip implant manufacturer Stryker reveals that certain sizes of their LFIT V40 Hip implant may rupture. The company received a higher than expected number of charges stating that the femoral head broke free of the stem that connects it to the thigh bone requiring revision surgery.
Stryker Orthopaedics is known for the development and manufacture of specialty surgical products and other medical supplies for health care around the world. The company has manufactured scores of medical devices and joint replacement products, most of which have been successful; however, some of the hip replacement devices have caused serious adverse events, injuring hundreds of patients.
The Stryker Rejuvenate Modular-Neck Stem and the ABG Modular-Neck Stem hip replacement devices were approved by the Food and Drug Administration (FDA) in 2008 and 2009. These devices are not typical "metal-on-metal" (MoM) as the necks are built of Cobalt and Chromium and the neck stems are layered with Titanium. Though the intention was to make it corrosion resistant, the two metals rubbed on each other at the connection and released toxic metal ions and debris. Stryker permanently recalled the Rejuvenate Modular and ABG II Modular-Neck Hip Stems in 2012 and stopped all global sales and production of these components. Another hip stem model, the Accolade TMZF, also caused problems and was recalled in 2009, 2011, and 2013.
Stryker identified eight sizes of LFIT V40 femoral heads that may cause complications and informed health centers to alert the patients. The FDA will decide in a month or two whether to consider a recall for the product or not. An earlier Class II recall by FDA in 2016 affected more than 42,500 Stryker hip replacement products.
Cases against Stryker hip replacement systems are consolidated as a part of multidistrict litigation, MDL No. 2768 ( In Re: Stryker LFIT V40 Femoral Head Products Liability Litigation) in the United States District Court for the District of Massachusetts before U.S. District Judge Indira Talwani.
Court Orders Hernia Mesh and Tissue Sample Preservation

The Federal Court issued the 11th Practice and Procedure Order on May 29 involving Ethicon Inc.'s Physiomesh Lawsuit. The order requires the hernia mesh and tissue samples to be preserved from plaintiffs who have been or will be requiring surgical removal and have a case pending in the proceeding.
The order also demands plaintiffs to inform their healthcare provider that any mesh or tissue removed must be preserved and sent to Steelgate, Inc., biomedical specimen storage, and management company. A ‘Preservation Notice’ has to be sent whenever the mesh removal process takes place. The further shipment of the explanted samples will be arranged by Steelgate, in Florida, which will act as a third-party storage unit for such materials.
In May 2016, Ethicon announced the withdrawal of its Physiomesh Flexible Composite Mesh from the worldwide market, considering reports that the device led to a lot of revision surgeries when used in laparoscopic ventral hernia repair.
The mesh products named in the lawsuits contain polypropylene material, which can easily break, shrink or wear off. This often leads to device migration, organ perforation, infection, and several other complications. Lawsuits allege the defective design of the hernia mesh often required the patients to undergo additional correction surgeries.
Over 900 Hernia Mesh lawsuits are pending against Ethicon; similar cases are consolidated as a part of multidistrict litigation MDL No. 2782 ( In Re: Ethicon Physiomesh Flexible Composite Hernia Mesh Products Liability Litigation) before the U.S. District Judge Richard Story in the Northern District of Georgia. The MDL is steadily moving forward with respect to the legal proceedings.
J&J to Face 22 Ovarian Cancer Claims; Imerys Opts Out

The Jury has selected 22 cases for a trial expected to convene on June 6 in St. Louis, Missouri, State Court, wherein the plaintiffs allege that Johnson & Johnson sold them cancer-causing talcum powder.
Johnson's Baby Powder, one of the most popular products containing talcum powder, is linked to increasing a woman's risk of ovarian cancer if she uses it regularly in the genital area. In a few cases, the cancer tissue was studied using an electron microscope and was found to have talc in it, which supported the claim that the cancer was caused by the body powder and increases the talc-related cancer risk.
Imerys SA settled the ovarian cancer claims by these 22 women for at least $5 million. The talc supplier faces 9000 claims stating the asbestos-laden talc supplied to J&J caused mesothelioma and ovarian cancer. This leaves J&J and its consumer-products unit as the only defendant in the case for not warning consumers about the talc cancer risk. However, Imerys maintains a firm denial regarding the presence of cancer-causing agents in the talcum powder. Thousands of lawsuits filed all over the nation allege the baby powder use for feminine hygiene purposes led to ovarian cancer in several women; many have concluded in multimillion verdicts against the talc manufacturer.
Similar lawsuits filed against J&J in state courts in California, New Jersey, and Delaware are consolidated for pre-trial proceedings in the U.S. District Court for the District of New Jersey MDL No. 2738 (In Re: Johnson & Johnson Talcum Powder Products Marketing, Sales Practices, and Products Liability Litigation), presided over by Hon. Freda L. Wolfson, U.S.D.J./ Hon. Lois H. Goodman, U.S.M.J.
Lead and Liaison Counsel Appointed for 3T Heater-Cooler MDL

The U.S.District Judge presiding over the 3T Heater-Cooler Systems pre-trial proceedings appointed a group of plaintiffs' attorneys to exhibit various leadership roles in taking the federal multidistrict litigation (MDL) forward.
The case management order issued by Judge Jones on May 31, stated one attorney should serve as lead and liaison counsel and an executive committee comprising six additional attorneys be made for taking timely actions during the litigation proceedings. However, each individual plaintiff’s own lawyer will be liable for deadlines and establish causation in the separate cases.
Stockert Heater-Cooler System (3T) is manufactured by LivaNova PLC (formerly Sorin Group Deutschland GmbH) and provides temperature-controlled water to heat exchanger devices (cardiopulmonary bypass heat exchangers, cardioplegia heat exchangers, and thermal regulating blankets) to warm or cool a patient during cardiopulmonary bypass procedures lasting six (6) hours or less. It is a Class II medical device that was approved by the U.S. Food and Drug Administration (FDA) via a 510K process in 2006. It is also called Sorin 3T.
Product liability cases continue to be reviewed by lawyers for open-heart surgery patients who suffered nontuberculous mycobacterium (NTM) infections due to exposure to infected 3T Heater-Coolers. Similar cases filed throughout the federal court system have been centralized under multidistrict litigation MDL No. 2816 (In Re: Sorin 3T Heater-Cooler System Products Liability Litigation No. II), before U.S. District Judge John E. Jones III in the Middle District of Pennsylvania.
Jury Sides the Defendant in the Second Bard IVC Filter Trial

The second bellwether trial for C.R. Bard's Eclipse IVC Filter ended in the defendant's favor on June 1, 2018, as the Arizona jury found that adequate warning was provided to doctors regarding the possible adverse effects of the IVC filter.
An Inferior vena cava filter (IVC filter), earlier popularly known as Greenfield filter, is a medical device implanted in the inferior vena cava just below the kidneys to capture blood clots, preventing them from reaching the heart and lungs, thereby, safeguarding against life-threatening pulmonary emboli (PE). IVC filters were cleared for use through the 510(k) process since 1976 however, in 2010 the FDA issued a device safety communication after reviewing more than 900 adverse events related to the devices over a period of five years C.R. Bard, Inc. and Bard Peripheral Vascular, Inc. (collectively, Bard) and Cook Incorporated, Cook Medical LLC, and William Cook Europe ApS (collectively, Cook) are the main manufacturers of retrievable IVC filters. Other manufacturers include Argon Medical Devices, Cordis Corporation, Rex Medical, Johnson & Johnson, ALN, B. Braun Medical, and Rafael Medical.
The verdict was given out in the U.S. District Court for the District of Arizona presided over by Judge David G. Campbell. The plaintiff was implanted with Bard IVC Eclipse Filter in 2010 to treat recurrent deep vein thrombosis and five years later she developed arm pain and headaches.
On further probe, it was discovered that the filter broke and one of the pieces blocked her right pulmonary artery, which could not be removed by surgeons.
The first bellwether against C.R.Bard concluded with $3.6 million as compensatory and punitive damages to the plaintiff. There are more than 3,800 IVC Filter lawsuits against Bard for not warning about the defective design of the filter. The cases were centralized in 2015 to coordinate the pre-trial proceedings under MDL 2641 (in re Bard IVC Filters Products Liability Litigation) before U.S. District Judge David Campbell.
Temporary Ban on Vaginal Mesh Use in the U.K.

The U.K. Department of Health and Social Care and National Health Service (NHS) England has decided to halt the use of vaginal mesh for treating stress urinary incontinence, acting on the request put forth by the Independent Medicines and Medical Devices Safety Review on July 10, 2018.
Transvaginal mesh is a surgical net-like implant, manufactured by various companies such as Ethicon, C.R. Bard, American Medical Systems, Boston Scientific, Coloplast, Cook Medical, Neomedic. It is used in the form of a sling to treat stress urinary incontinence (SUI) and Pelvic organ prolapse (POP) in women since the 1990s. Abdominal hernias have also been treated with a surgical mesh since the 1950s. The insertion of this mesh or a bladder sling through the vagina is known as a transvaginal mesh. The Food and Drug Administration (FDA) approved the first surgical mesh specifically designed for SUI in 1996. Later, in 2004, the FDA approved the first surgical mesh specifically for use in POP.
The temporary ban on the vaginal mesh will stay until March 2019, but the review board asserts that the devices should be back into the UK market only after assuring that a substantial amount of the risks can be curbed. In her press release statement, the review board chairperson Baroness Julia Cumberlege told, “We strongly believe that mesh must not be used to treat women with stress urinary incontinence until we can manage the risk of complications much more effectively. We have not seen evidence on the benefits of mesh that outweighs the severity of human suffering caused by mesh complications. I have been appalled at the seriousness and scale of the tragic stories we have heard from women and their families. We have heard from many women who are suffering terribly.” The ban would be removed after a detailed inspection and accreditation of all specialist centers that conduct stress urinary incontinence mesh procedures.
Transvaginal mesh lawsuits continue to be filed in the U.S. District Courts against manufacturers including Ethicon, C.R. Bard, Boston Scientific over design flaws causing severe internal injuries in several women. All of the TVM multidistrict litigation in the U.S. over failure to warn allegation is overlooked by Judge Joseph R. Goodwin in the Southern District of West Virginia.